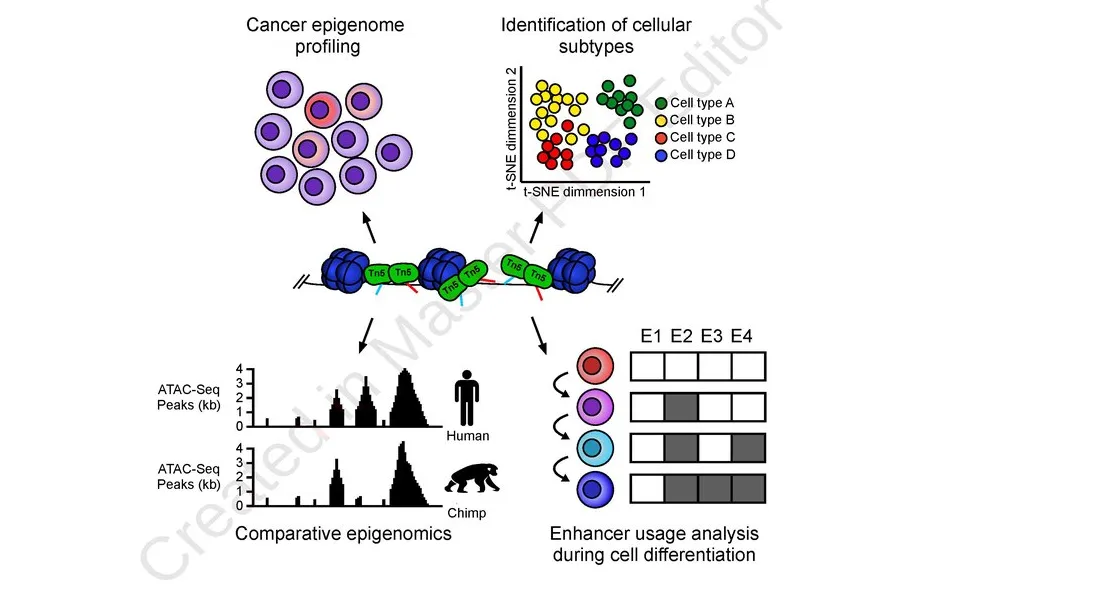
单细胞技术的发展使得我们能够更深入地探究细胞的个体差异性和异质性。通过整合多组学数据,例如单细胞 ATAC-seq 和 RNA-seq,我们能够揭示细胞在转录调控中的复杂关系,解析基因表达和染色质开放性的联系,有助于理解细胞功能的精细调控机制。
让生信分析更简单高效,点击图片访问【生信圆桌】获取生信云服务器;下单填写优惠码 tebteb 立省200元

单细胞 ATAC-seq 与 RNA-seq 的整合
1.背景简介
ATAC-seq(Assay for Transposase-Accessible Chromatin using sequencing)与 RNA-seq 分别用于表观遗传和转录组数据的获取,前者反映了染色质开放性,后者则揭示了基因表达水平。二者的整合有助于识别调控元件与其靶基因之间的关系,提供关于基因表达调控的全面视图。
2.数据预处理与标准化
整合 ATAC-seq 和 RNA-seq 数据需要进行质量控制、归一化等处理,以减小因技术差异引起的偏倚。常用的方法包括对数据进行 Log 转换、归一化和滤波等。
3.整合方法与算法
在整合分析中,最常用的方法包括多层次的矩阵分解和深度学习算法,此外,CCA(Canonical Correlation Analysis)、MNN(Mutual Nearest Neighbors)、Harmony 等也是常用的整合方法。它们能够找到相似的细胞群体,从而帮助识别调控网络和细胞类型特异的调控因子。
4.常见挑战和应对策略
ATAC-seq 数据通常噪音较高,且信号分辨率低,数据整合前需使用滤波、批次校正等处理,减少误差累积的影响。为克服细胞异质性造成的偏差,常采用 Harmony 或 scTransform 等算法进行校正。此外,单细胞整合往往面临大规模数据计算的挑战,因此需考虑云计算等高性能计算资源以提升处理效率。
跨平台整合分析的挑战与解决方案
1.数据异质性
不同平台产生的单细胞数据在分辨率、数据质量等方面存在差异,导致数据不一致。为解决这一问题,通常使用批次校正算法,如 Harmony、scTransform 等,以消除技术噪声,确保数据的可比性。
2.数据稀疏性
单细胞数据往往是稀疏的,尤其在低覆盖度的 ATAC-seq 中更为明显。稀疏数据会影响整合效果,因此可以通过使用深度学习模型(如 scVI)来对数据进行填补和增强,以提高数据整合的准确性。
3.标记细胞群体的难度
在整合不同平台数据时,通常需要准确地标记细胞类型以对齐相似的细胞群体。基于知识的标注工具(如 SingleR 和 CellTypist)能够通过比对参考数据来提供较为可靠的细胞类型标注。
4.算法性能与计算资源需求
大规模单细胞多组学数据的整合分析计算成本高,可通过使用云计算加速技术优化计算效率。当前,一些分布式计算框架和云服务(如 生信圆桌|生信云解决方案,利用分布式集群加速科研计算效率,减少用户成本)能够加速数据处理过程,提升算法的可扩展性。
应用与前景
整合单细胞 ATAC-seq 和 RNA-seq 数据不仅能够揭示细胞类型特异性的基因调控网络,还有助于肿瘤微环境、发育生物学、免疫学等领域的深入研究。这种整合分析正在逐步成为精准医学研究中的重要手段。