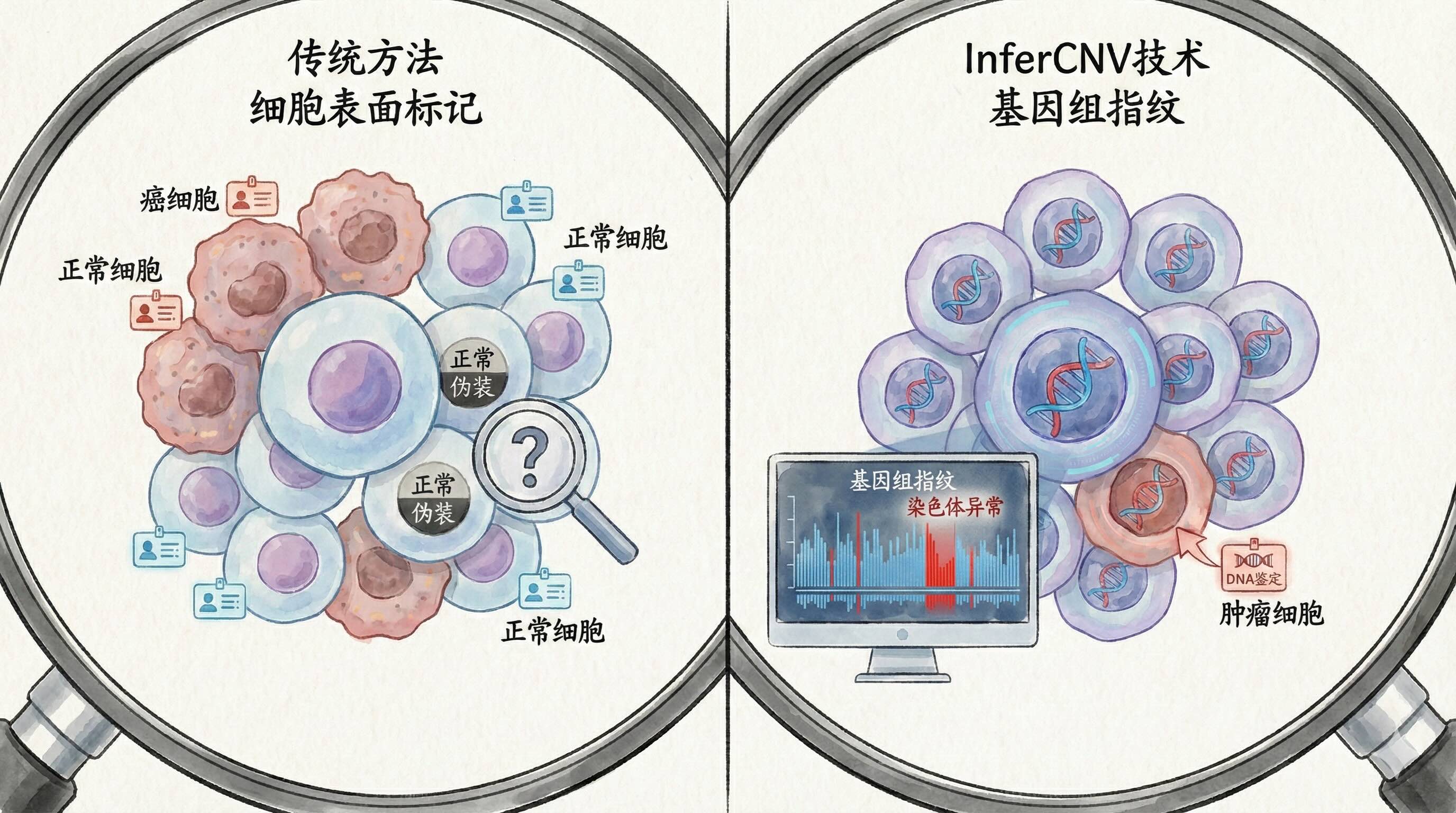
一项名为InferCNV的计算生物学技术登场了。它不看细胞的"外表",而是直接检查细胞的"基因组指纹"——就像警察通过DNA鉴定罪犯一样。这项技术能从单细胞测序数据中揪出那些携带染色体异常的肿瘤细胞,准确率令人惊叹。
一、肿瘤的"身份危机":为什么识别癌细胞这么难?
1.1 肿瘤不是"一块铁板"
我们常说"得了癌症",但其实肿瘤并非由单一类型的细胞组成。它更像一个复杂的"生态系统",里面既有疯狂增殖的癌细胞,也有被招募来的免疫细胞、血管细胞、成纤维细胞等。这些细胞混杂在一起,就像人群中混入了间谍,如何精准识别成了科学家面临的首要难题。
传统的批量测序技术就像对整个人群拍一张集体照——你只能看到平均的特征,却无法分辨每个人的独特面貌。而**单细胞RNA测序(scRNA-seq)**技术的出现,让我们能够给每一个细胞拍"单人照",细致入微地分析它们的基因表达状态。
1.2 "长得像"不代表"是同一种"
但新问题又来了:仅凭基因表达来判断细胞身份并不可靠。比如,一个正在快速分裂的正常上皮细胞,其基因表达模式可能和肿瘤细胞极为相似;反之,处于休眠状态的肿瘤干细胞可能伪装得像正常干细胞。这就好比一个正在健身的普通人,和一个假装休息的特工,仅从表情动作很难区分。
因此,我们需要找到一个更本质的标准——染色体拷贝数变异(CNV)。
![生信分析跑不动?试试稳定高性价比的生信云 → [www.tebteb.cc]](https://www.tebteb.cc/upload/%E7%94%9F%E4%BF%A1%E5%9C%86%E6%A1%8C-kgzw.png)
二、肿瘤的"基因组指纹":什么是拷贝数变异?
2.1 正常细胞的染色体"家底"
人类的正常细胞拥有23对染色体,每条染色体上携带着成千上万的基因。这些基因的拷贝数是固定的——大部分基因都是"两份"(一份来自父亲,一份来自母亲)。这种稳定的基因组结构维持着细胞的正常功能。
2.2 肿瘤细胞的"基因组大地震"
而癌细胞在恶变过程中,染色体常常发生大规模的重排:
扩增(Gain):某段染色体区域被复制了多次,导致上面的基因拷贝数增加到3份、4份甚至更多
缺失(Loss):某段染色体区域丢失,导致基因拷贝数减少到1份或完全消失
这种大范围的染色体异常在正常细胞中极为罕见(除了少数特殊情况如免疫细胞的基因重排),因此成为了识别肿瘤细胞的"金标准"特征。
2.3 基因剂量效应:从DNA到RNA的逻辑链
这里有个关键原理:基因拷贝数的改变会影响基因的表达量。
如果7号染色体扩增了(变成3份或4份),那么这条染色体上的基因,其mRNA转录水平通常也会相应上升
如果10号染色体缺失了(只剩1份或0份),该区域基因的mRNA水平就会下降
这种现象被称为"基因剂量效应"。虽然单个基因的表达受很多复杂因素影响,但当我们把一整条染色体臂上数百个基因的表达量平均起来看,拷贝数带来的整体"基线偏移"就会显现出来——这正是InferCNV的理论基础。
三、InferCNV:一把"基因组手术刀"
3.1 它是什么?
InferCNV是由美国布罗德研究所(Broad Institute)开发的一款开源生物信息学软件。它的核心能力是:从单细胞转录组数据中推断出每个细胞的染色体拷贝数变异情况。
简单来说,它不直接测量DNA(那需要单细胞全基因组测序,成本高昂),而是通过巧妙的数学算法,从RNA测序数据中"逆推"出DNA的拷贝数状态。
3.2 工作原理:五步"侦探术"
InferCNV的分析流程可以类比为一场精密的刑侦工作:
第一步:建立参考基线 首先需要找到"良民"——那些确定是正常细胞的群体(如样本中的免疫细胞、内皮细胞)。计算这些正常细胞中每个基因的平均表达量,作为"正常基线"。
第二步:计算表达偏差 对于每个待检测的细胞,将其基因表达量减去正常基线。这个差值(残差)反映了该细胞相对于正常状态的偏离程度。
第三步:染色体平滑处理 单个基因的表达充满随机噪声,但拷贝数变异影响的是整片染色体区域。因此,算法会沿着染色体的物理位置,对相邻的约100个基因进行移动平均平滑——就像用滤镜去掉照片噪点,让真正的信号浮现出来。
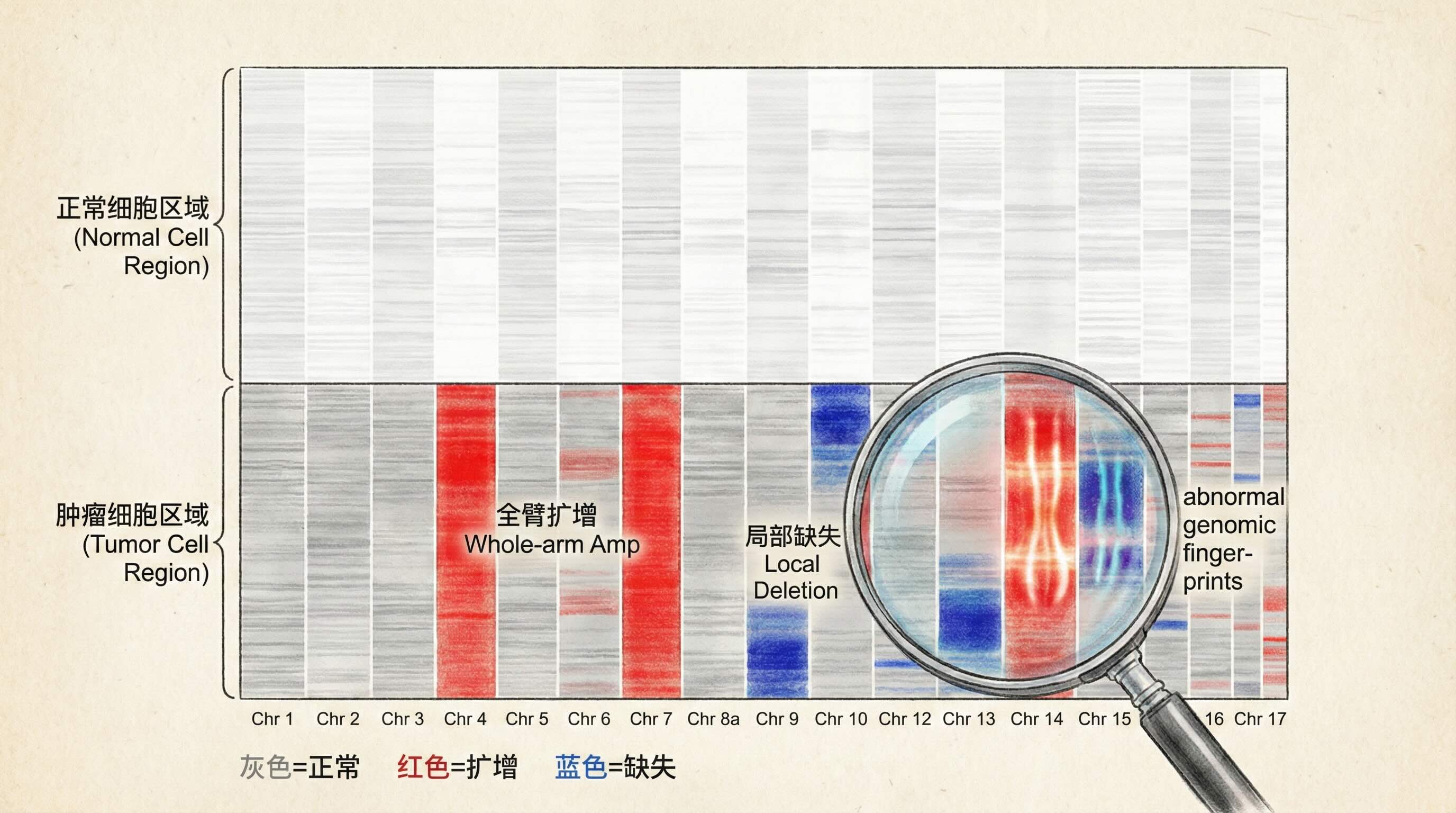
第四步:生成热图 最终输出一张彩色热图:
红色区域:表示该染色体片段的基因表达高于正常,对应拷贝数扩增
蓝色区域:表示基因表达低于正常,对应拷贝数缺失
白色/灰色:表示正常
第五步:智能预测 高级版本还会使用隐马尔可夫模型(HMM)和贝叶斯统计,自动判断每个染色体区域的拷贝数状态(如"正常2份""扩增3份""缺失1份"),并评估预测的可信度。
3.3 一张热图胜千言
当你看到InferCNV的输出热图时,正常细胞区域应该是平淡的白灰色(表示没有明显的染色体异常),而肿瘤细胞区域则会出现醒目的红蓝条带——有的整条染色体都是红色(全臂扩增),有的某个区域是深蓝(局部缺失)。
这种视觉上的对比一目了然,就像在混乱的人群中,那些携带"异常基因组指纹"的肿瘤细胞立刻暴露了身份。
四、实战应用:科学家如何用InferCNV破案?
4.1 案例:胶质母细胞瘤的克隆追踪
胶质母细胞瘤(GBM)是一种极其凶险的脑瘤。研究人员利用InferCNV分析患者的单细胞数据后发现:
几乎所有肿瘤细胞都携带7号染色体扩增和10号染色体缺失
但其中一部分细胞额外获得了19号染色体缺失
这说明肿瘤在进化:最初的"祖先克隆"只有7+/10-变异,后来演化出了带有额外突变的"子代克隆"。通过InferCNV的聚类树分析,科学家能够重建肿瘤的进化家谱,了解哪些变异是早期驱动事件,哪些是后期获得的。
4.2 无参考样本怎么办?
有时候,样本中根本没有明确的正常细胞(比如纯肿瘤的穿刺活检)。InferCNV也能应对——它会用所有细胞的平均值作为基线。
此时,如果90%是肿瘤细胞、10%是正常细胞,热图上会出现"镜像"效果:真正的正常细胞(10%)看起来像是"反向异常"的,而占主导的肿瘤细胞(90%)则呈现出典型的CNV模式。研究人员需要结合医学知识判断——通常基因组越"混乱"的那一组才是真正的肿瘤。
五、新手入门:从零到一的实践指南
5.1 你需要准备什么?
测序数据:单细胞RNA测序的原始计数矩阵(未标准化的UMI counts)
细胞注释文件:标明哪些细胞是T细胞、哪些是内皮细胞等
基因位置文件:每个基因在染色体上的物理坐标
计算资源:建议64GB内存以上的服务器,因为数据处理非常消耗资源
5.2 避坑指南:新手最常犯的三大错误
错误1:参数设置不当 10x Genomics平台的数据很稀疏,基因表达阈值应设为0.1;而Smart-seq2数据更深,可以设为1。用错参数会导致要么过滤掉所有信号(热图全白),要么噪声太多(一片杂乱)。
错误2:参考细胞选择不纯 如果你把一些肿瘤细胞误标为"正常参考",那么算法就会用错误的基线,导致真正的肿瘤信号被掩盖。务必使用那些明确无疑是正常的细胞类型(如免疫细胞)作为参考。
错误3:基因组版本混乱 如果测序比对时用的是hg38参考基因组,但InferCNV用的基因位置文件是hg19,那么基因的物理顺序就会错乱,平滑算法完全失效,输出的热图就是一团噪声。
六、未来展望:InferCNV的进化与竞争
6.1 新工具的挑战
近年来,几款新工具开始挑战InferCNV的地位:
CopyKAT:无需指定参考细胞,能自动寻找正常细胞群体,适合"盲测"场景
Numbat:结合SNP等位基因信息,不仅能检测拷贝数变化,还能发现"拷贝数中性的杂合性缺失"(CN-LOH)——一种InferCNV无法检测的隐秘突变
不过,InferCNV凭借其成熟的算法、丰富的文档和广泛的用户群体,依然是目前的行业标准。
6.2 空间转录组的新战场
随着Visium等空间转录组技术的兴起,InferCNV被用来绘制"肿瘤克隆的空间地图"。科学家可以回答诸如:"携带7号染色体扩增的侵袭性克隆,是否更倾向于分布在肿瘤与正常组织的交界处?"这为理解肿瘤的空间生态学打开了新窗口。
结语
InferCNV就像一把"基因组手术刀",让科学家能够精准剖析肿瘤细胞的身份。它不依赖细胞的表面伪装,而是直接检查最本质的基因组特征——染色体拷贝数变异。通过巧妙的算法设计,它将高噪声的单细胞转录组数据转化为清晰的CNV图谱,为癌症研究提供了不可或缺的工具。
对于科研人员而言,掌握InferCNV不仅是学会操作软件,更是理解"基因剂量效应"原理、精心设计实验流程、正确解读生物学信号的系统性训练。随着单细胞技术向更大规模和更多维度发展,InferCNV及其衍生方法,必将在精准医疗和肿瘤演化研究中发挥更加重要的作用。
当下一次你听到"单细胞测序""肿瘤异质性"这些术语时,不妨想象一下那张红蓝交织的InferCNV热图——那是科学家用数学和计算机为我们打开的一扇窗,透过它,我们能看见癌细胞最隐秘的基因组真相。
InferCNV
单细胞RNA测序
拷贝数变异
CNV分析
肿瘤细胞识别
scRNA-seq
染色体异常检测
基因剂量效应
肿瘤异质性
生物信息学工具
癌症基因组学
恶性克隆
HMM预测
单细胞测序分析
肿瘤微环境